Phylogenetics [1]
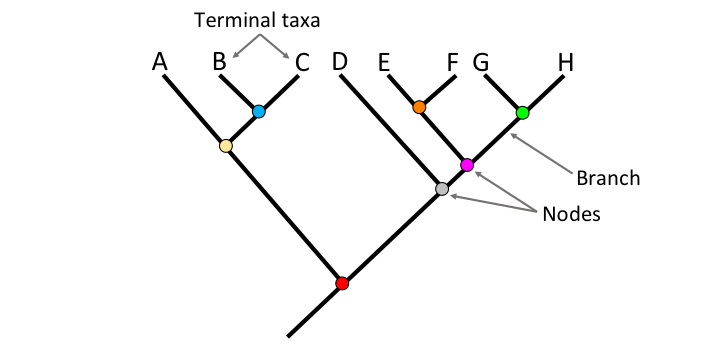
Phylogenetics is the study of biological relation based on evolutionary similarities and differences. Phylogenetic findings are often displayed in tree diagrams where each branch can represent the evolutionary history of changes a gene or species has undergone over time. Nodes are a branching point where a common ancestor diverged down more than one genetic path. The root of the tree symbolizes the common ancestor of all of the biological units on the tree. A clade is a grouping of closely related branch tips along with their common ancestor. In the past, trees were constructed by observing morphological differences among species, but with the advancement of modern technology, we are now capable of making much more precise trees by comparing and contrasting DNA and protein sequences.
Phylogeny Methods [2]
Maximum Likelihood takes every possible tree into account to decide which one is the most likely. It evaluates the probability of one sequence changing to another for every single position in order to determine which chain of events has the highest chance of being the true tree. This method requires a monstrous amount of calculations, but if you have sufficient data and the computational means to use maximum likelihood, it is arguably the most accurate tree construction method.
Neighbor Joining uses an algorithm to determine which branch lengths are the shortest for each level of the tree. After finding the pair with the lowest sum, the algorithm considers that pair closely related, and moves on to the next level until the tree is completed. This method can be completed much quicker than maximum likelihood and does not require as much data, but it is virtually unidirectional, so the output is always just one tree, making this method not quite as reliable as maximum likelihood.
Minimum Evolution aims to keep the total branch length as small as possible. Minimum evolution and neighbor joining produce comparable trees in most cases. This method can be susceptible to neglecting factors such as homoplasy, reversals, and convergent evolution. These errors can become more of a problem for tree groupings that go relatively deep into evolutionary history.
Neighbor Joining uses an algorithm to determine which branch lengths are the shortest for each level of the tree. After finding the pair with the lowest sum, the algorithm considers that pair closely related, and moves on to the next level until the tree is completed. This method can be completed much quicker than maximum likelihood and does not require as much data, but it is virtually unidirectional, so the output is always just one tree, making this method not quite as reliable as maximum likelihood.
Minimum Evolution aims to keep the total branch length as small as possible. Minimum evolution and neighbor joining produce comparable trees in most cases. This method can be susceptible to neglecting factors such as homoplasy, reversals, and convergent evolution. These errors can become more of a problem for tree groupings that go relatively deep into evolutionary history.
Constructing a PTK7 Phylogenetic Tree
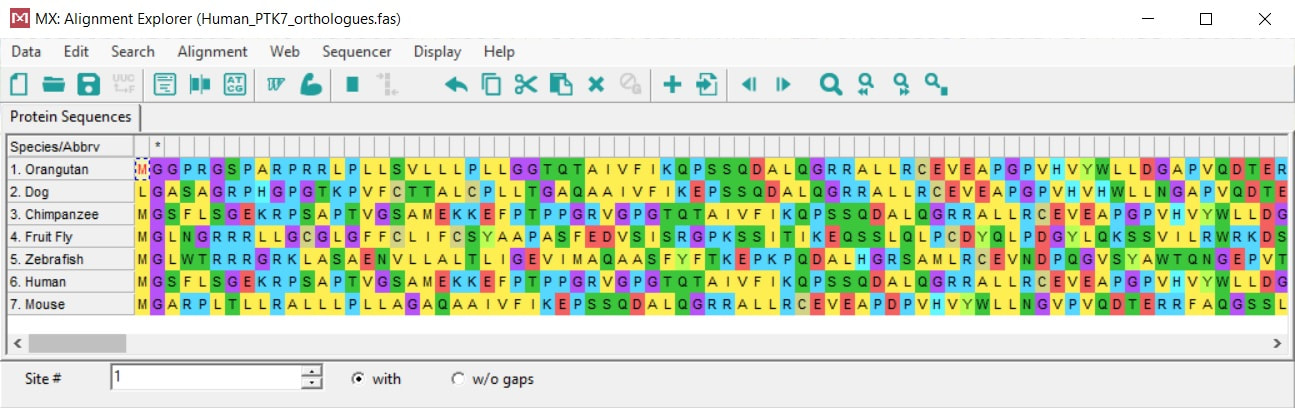
Above is the alignment of amino acid sequences for PTK7 orthologs using MEGAX.
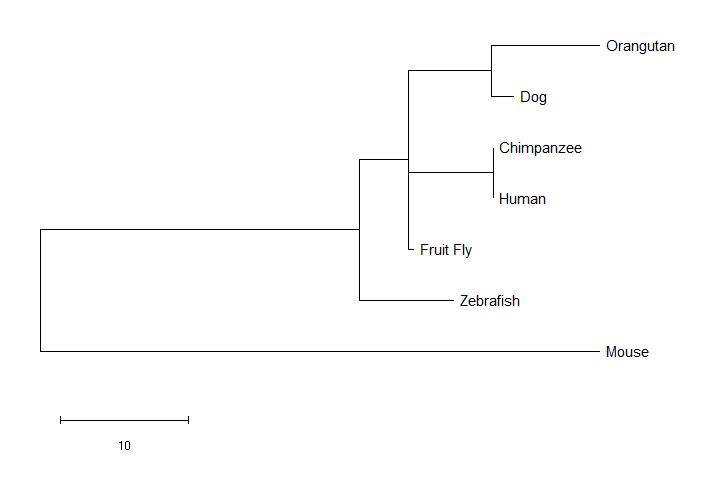
PTK7 Maximum Likelihood Tree
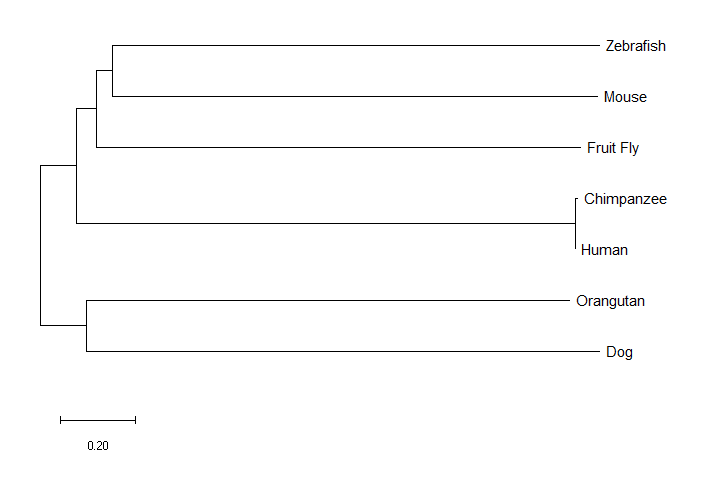
PTK7 Neighbor Joining Tree
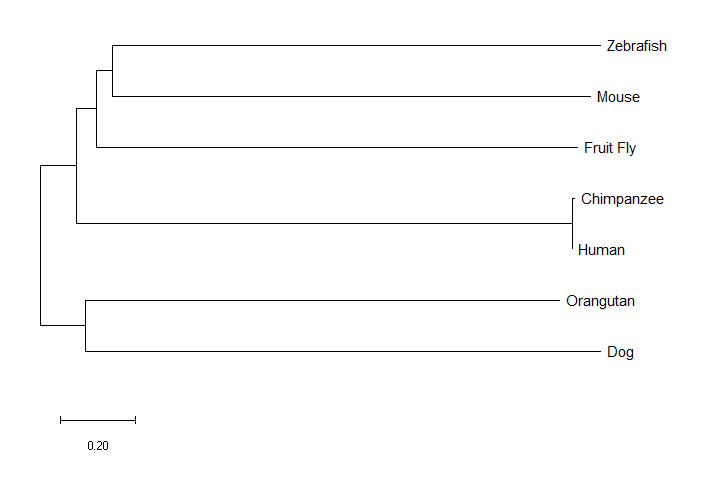
PTK7 Minimum Evolution Tree
Conclusion
The neighbor joining tree and the minimum evolution tree were identical, which isn't all that surprising, as it was noted in the literature that those two methods tend to have similar outcomes [2]. The maximum likelihood tree was different. For example, the mouse was an outgroup in the maximum likelihood tree. As I expected, the chimpanzee was most closely related to humans, but I was a bit surprised that all three methods found the orangutan ortholog to be more closely related to the dog than to humans or the chimpanzee.
References:
[1] https://www.nature.com/subjects/phylogenetics
[2] http://guava.physics.uiuc.edu/~nigel/courses/598BIO/498BIOonline-essays/hw2/files/hw2_li.pdf
Header image from: https://www.amazon.com/Charles-Species-Evolution-Drawing-Stationery/dp/B0742FHGZ9
All trees were made using MEGAX.
[1] https://www.nature.com/subjects/phylogenetics
[2] http://guava.physics.uiuc.edu/~nigel/courses/598BIO/498BIOonline-essays/hw2/files/hw2_li.pdf
Header image from: https://www.amazon.com/Charles-Species-Evolution-Drawing-Stationery/dp/B0742FHGZ9
All trees were made using MEGAX.
This web page was produced as an assignment for Genetics 564, an undergraduate capstone course at UW-Madison.
